




研制新型强效止痛药是一场对抗副作用的持久战,尤其是对抗药物成瘾。James Mitchell Crow报道。
对于一位发现改变了世界的化学家来说,弗里德里希·威廉Sertürner这个名字令人惊讶地很少有科学家知道。1783年出生于德国北部,这位才华横溢的年轻化学家是一名药剂师,1804年,他在研究罂粟的提取物时发现了吗啡——有史以来第一个被分离出来的天然产物之一。
通过发现一种从粗鸦片树脂中分离出纯止痛药的方法,Sertürner为第一个剂量控制的疼痛药物铺平了道路。200多年后,吗啡仍然是我们所知道的最有效的止痛药之一,它仍然是当今医学上控制剧烈疼痛的重要工具——尽管这种药物有臭名昭著的副作用。
到了Sertürner的时代,鸦片的成瘾性已经广为人知。已知的第一次使用这种植物可以追溯到公元前3400年,当时美索不达米亚的苏美尔人已经在种植罂粟,他们将其命名为“欢乐植物”。
不幸的是,鸦片的成瘾性影响到了吗啡。Sertürner自己可能已经屈服于化合物的阴险的控制。众所周知,他在自己身上进行了测试,当代对他晚年性格变化和“情绪紊乱”的描述使许多人得出结论,Sertürner成为了一个吗啡成瘾者,也许是世界上第一个。
Sertürner在1841年去世后不久,化学家们开始寻找一种阿片类化合物,这种化合物既保留了吗啡的止痛特性,又没有有害的副作用。这些因素包括耐受性(重复服用药物后药效下降)、成瘾、便秘和呼吸抑制(导致服用过量患者死亡的副作用)。这是一场曲折的探索,与我们对体内药物受体相互作用的不断发展的理解相一致。过去12个月发表的两篇论文表明,我们终于接近我们的猎物了。
迫切需要
迫切需要更好的阿片类药物。如今,五分之一的成年人目前处于中度至重度慢性疼痛之中。持续疼痛带来的失业、抑郁和人际关系破裂的几率明显更高。
由于缺乏有效的药物来帮助这些患者,20世纪80年代中期,医生开始为慢性疼痛开出强效阿片类药物,如羟考酮(oxycodone),它的结构与吗啡非常相似。这些化合物强大的止痛作用使它们成为短期控制急性疼痛的特殊药物,但它们的副作用使它们成为慢性疾病的高风险选择。
“羟考酮在治疗慢性疼痛方面存在问题的第一个证据来自于一项研究梅奥疼痛诊所1981年发表的研究澳大利亚纽卡斯尔亨特疼痛诊所(Hunter pain Clinic)的疼痛医学专家马克•罗素(Marc Russo)说。“这说明了痛苦的力量,我们接受了它有好有坏——我们没有说‘这还不够好’。”
由此引发的健康危机被称为“阿片类药物流行病”,这种危机在许多国家都发生过,但在美国尤为严重。到2014年,近200万美国人被归类为处方阿片类药物成瘾者。那一年,近2万美国人死于处方阿片类药物过量。
2016年3月,美国疾病控制中心(CDC)发布了新的阿片类药物处方指南为了控制疫情但潜在的问题仍然存在:迫切需要一种安全有效的药物来缓解数百万慢性疼痛患者的痛苦。
英雄还是恶棍?
一个多世纪前,沿着这条道路迈出的第一步并没有带来多大的成功。最早合成的吗啡衍生物之一是它的乙酰化形式。二乙酰吗啡于1873年首次被制造出来,但在1896年由拜耳制药公司的化学家独立重新合成,该公司很快就开始以“海洛因”的品牌名出售它,因为它据称对使用者产生了巨大的影响。
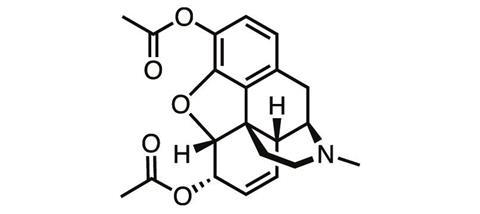
美国杜兰大学和新奥尔良退伍军人事务部的药物化学家詹姆斯·扎迪纳说:“海洛因刚问世时,被吹捧比吗啡更好,因为它不会上瘾。”“我们知道结果有多好。扎迪纳补充说:“海洛因优越的论点背后的‘逻辑’是,它的化学纯度比吗啡更高,或者药效更强,但这两者都不相关。”
直到20世纪30年代,我们还没有任何可靠的动物实验来评估药物的成瘾性。在此之前,成瘾一直被认为是人类特有的现象。佛罗里达耶鲁大学灵长类生物学实验室对黑猩猩进行了注射吗啡的实验,初步揭示了成瘾的潜在生理基础。依赖药物的黑猩猩会开始把研究人员拖到注射室进行下一次注射。现代的“条件性地点偏好”动物测试在啮齿动物中寻找类似的行为。给老鼠服用具有成瘾性的药物后,老鼠会在给药的地方待更长的时间。
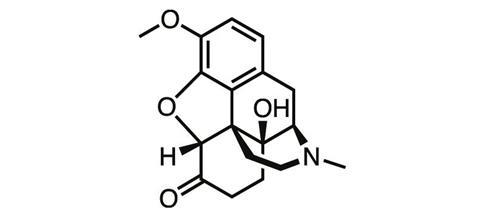
随着海洛因的真实性质越来越明显,化学家们又开始研究罂粟,这次他们试着用罂粟的另一种天然产物——大麻碱来碰碰运气。1916年,法兰克福大学的德国化学家马丁·弗罗因德和埃德蒙·斯派尔通过一系列氧化和还原步骤,从thebaine合成了羟考酮。但阿片类药物的耐受性、成瘾性和滥用性仍然存在。
它们仍然存在于合成阿片类药物中,如1939年发现的哌嗪(哌啶)和芬太尼(1960年),后者在添加各种结构修饰之前,将吗啡分子剥离回含氮的六元“哌啶”环。
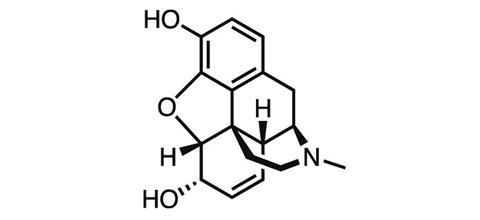
但在化学研究的同时,人体-阿片类药物相互作用的生物学也在被探索——在20世纪70年代,我们似乎终于找到了解决副作用问题的方法。1977年,阿伯丁大学的汉斯·科斯特利茨(2016年诺贝尔物理学奖得主迈克尔·科斯特利茨的父亲)和同事们展示了人体自己产生镇痛“内源性阿片类药物”它激活的不是一种,而是一系列阿片受体亚型。
这一发现提出了一种诱人的可能性,即如果我们能够研制出一种药物,可以选择性地针对四种阿片类受体亚型中的一种——也许是通过使用或改造内源性阿片类药物中的一种——我们就可以将镇痛作用与副作用分开。
但事实并非如此。吗啡本身,以及芬太尼,被证明是非常选择性的一个亚型,μ-阿片受体。只击中这种亚型似乎会同时引发好的和坏的影响。希望消失了。阿片类药物行为的两个方面似乎密不可分。
扎迪纳说:“我认为我们认为受体有点像图画:它们是漂亮的、静态的小圆圈和正方形的洞,被小圆圈和正方形塞进去。”“我们认为任何撞击受体的分子都会产生相同的反应,引发相同的反应。现在我们知道情况要复杂得多。
二十年的工作
最初的迹象之一是阿片类药物研究的受体亚型章节并不是故事的结尾1997年.在Kosterlitz的工作大约20年后,Zadina和他的团队研究了已知的内源性阿片类物质和已知的阿片类受体,并注意到一些缺失的东西:没有一种内源性阿片类物质对μ-阿片类受体表现出任何很强的选择性,而吗啡对μ-阿片类受体表现出如此强的亲和力。没有一种内源性化合物看起来像那种受体的天然激动剂。深入研究大脑后,研究小组发现了内啡素-1,这是第一个对μ受体的选择性与吗啡相当的内源性阿片类物质。
从一开始,内啡肽-1似乎在体内引起了与任何其他阿片类物质不同的反应。扎迪纳回忆说:“即使是使用原始的内源性化合物,我们也可以看到,它在镇痛方面优于条件部位偏好(其成瘾性的动物试验),在抑制呼吸方面也更好。”“这就是为什么我在接下来的20年里用这些化合物制造结构类似物的原因。”
扎迪纳在工作中有三个目标。他说,这种天然化合物是一种短肽,“在血液中很快就会被消化掉”。该团队通过形成环状类似物来稳定结构,这是一种成熟的技术,用于抵御蛋白酶,否则蛋白酶会在体内代谢它们。
另外两个目标是最大限度地提高镇痛效果,使其达到吗啡的效果或更好,同时最大限度地减少副作用,特别是滥用的风险。2015年12月下旬,Zadina发表了这项工作的成果,一系列内胚素类似物在动物试验中表现良好,该团队现在正在为人类临床试验做准备。
在接下来的20年里,我用这些化合物制造结构类似物
詹姆斯·扎迪纳,杜兰大学
扎迪纳说:“我们发现了一种化合物,它能产生非常低的呼吸抑制,镇痛时间比吗啡长得多,耐受性也小得多。”但扎迪纳实验室的真正重点是彻底测试上瘾特性。最复杂的潜在滥用动物测试之一是将一只大鼠连接到静脉导管上,当动物按下一个按钮时,静脉导管就会输送吗啡。扎迪纳说,静脉注射吗啡后,老鼠会“开始疯狂地推杆”。“它们不会显著增加我们化合物的压杆。我们认为这是最引人注目的实验,不过只有在人体上进行试验才能知道结果。”
扎迪娜的先导化合物有这么大的副作用,背后是什么?扎迪纳说:“过去的想法是,任何击中该受体的药物都会产生几乎相同的效果。”“现在我们知道,不同的结构会产生不同的反应模式。“激动剂在受体结合袋内采用的不同构象触发了不同的信号模式。“我们希望找到一套更好的镇痛模式,如果你能优先考虑这一途径,而不是导致副作用的途径,你可能会得出更好的结果。”
这一概念被称为偏激效应,是g蛋白偶联受体(GPCR)生物化学的一个方面,杜克大学的罗伯特·莱夫科维茨和美国斯坦福大学的布莱恩·科比尔卡因此被授予2012年诺贝尔化学奖。科比尔卡在斯坦福大学的同事阿什什·曼格利克(Aashish Manglik)说:“人们正试图找到针对所有GPCRs的有针对性的药物,以更好地控制药物的作用。”
对于阿片类药物,也许最引人注目的结果是劳拉·博恩(Laura Bohn)的工作,她与莱夫科维茨合作,研究了吗啡对缺乏β-抑制素(一种GPCR调节蛋白)的小鼠的副作用。与对照组相比,这些小鼠表现出较强的镇痛作用、较低的药物耐受性、较低的便秘和呼吸抑制。
扎迪纳补充说,很少有人提到的是,这些老鼠在评估上瘾的“条件条件”测试中表现更差,所以寻找简单地避免β-抑制素信号的化合物不太可能是全部答案。他说:“我自己的偏见是,消除这种情况不太好。”“你可能只需要找到激动剂可能诱导的信号模式的正确组合。”
新希望
扎迪纳并不是唯一一个通过大脑而不是罂粟植物来寻找新的阿片类药物线索灵感的人。2012年,作为科比尔卡实验室的研究生,曼格利克和他的同事终于打破晶体结构这一切的核心蛋白质:μ-阿片受体。
曼格里克解释说:“我们主要是出于对GPCRs如何发挥作用的非常基础的科学兴趣,阿片受体是一个原型家族。”曼格里克最近离开了科比尔卡的实验室,在斯坦福大学建立了自己的团队。“但是GPCR结构生物学的真正希望和梦想是影响药物开发,”他补充道。“当我们第一次得到这种受体的照片时,我们决定与加州大学旧金山分校的布莱恩·绍切特合作,看看能否用它来寻找阿片受体的新分子。”
曼格利克、科比尔卡、绍切特和他们的公司后来成立了一个药物发现超级集团。利用Manglik的晶体结构,研究小组计算了300多万种化合物与μ-阿片受体结合的能力,得到了2500个命中。他们将这个列表缩小到几十个显示出最佳结合潜力的药物,这些药物与现有的阿片类药物具有非常不同的化学结构。肖切特说:“我们不想只是优化已经存在的化学反应。”“我们希望得到一种新的化学物质,它将赋予我们全新的生物学。德国埃尔兰根弗里德里希-亚历山大大学的Peter Gmeiner团队合成了这种主要化合物,然后在美国北卡罗来纳大学教堂山分校的Brian Roth实验室进行了测试。
结果是PZM21,这是一种新型化合物,迄今为止在动物实验中显示,在等效镇痛剂量下,它比吗啡表现出更少的呼吸抑制、便秘、耐受性或成瘾。Manglik说:“这种分子现在是一种药物的先导,我们还没有发现阻碍进一步开发的缺陷。”“对于药物开发来说,一种特定分子失败的概率相当高,所以我们会继续制造新版本,并寻找保留有利副作用的变体。”
“这是一项非常令人兴奋的研究,”扎迪纳说。“我不知道这会不会发生的化合物,”他补充道,并指出到目前为止,该团队只发表了有限的关于化合物成瘾性的动物试验数据。“但这很好,因为它强化了这样一个观点,即我们应该能够制造出区分好坏影响的激动剂。”同年,我们的论文和这篇论文发表,都强化了这一观点。”
Manglik表示同意。扎迪纳集团采取了非常理性的做法。它们有一些非凡的特性,其中一些数据非常令人兴奋。
曼格利克补充说:“每一种具有这些有趣性质的化合物都是深入研究系统的另一种方式。”即使从长远来看,这些特殊的有偏见的激动剂不能作为药物先导,“它们是理解生物学的先导化合物,可以开始深入研究信号,并真正理解发生了什么”。基于这一认识,新的药物线索将会出现。
正如罗素所指出的,这一次,在向世界发布一种新的阿片类药物之前,我们希望非常确定。“它们需要再次进行比较,进行临床评估,在没有确定它们是否比我们现有的更好的情况下,不能把它们随意地交给医生。”
詹姆斯·米切尔·克罗是澳大利亚墨尔本的科学作家






暂无评论