




为什么这么多药物结构相似之处吗?如果你看的很多最畅销的和most-prescribed药物——比如sofosbuvir丙型肝炎治疗,抗高血压赖诺普利或哮喘的药物——他们有一些共同点:他们是人口functionalised,至少有一个环和一些保护胺或醇,并经常氟原子的散射。
答案的一部分是,药物发现化学家是有限的化学空间,可以通过已知的反应和访问可用的构建块。他们还需要考虑生物学,所以反应容忍基本氮原子是必须的因为他们特性几乎在每一个生物活性化合物。只使用现成的反应试剂不着火时暴露在空气中也是一大亮点,因为通常是不容易获得专业设备喜欢手套箱。
2014年的一项分析1显示,只有两种反应占超过一半的那些用于发现和开发药物的合成:酰胺形成和Suzuki-Miyaura交叉耦合。的这些贡献了这么多药物化学的原因是,他们是最健壮的反应,”说乔治- Keserű,他领导着一个药物化学组匈牙利科学院。
从看似简单的潜在曾获诺贝尔奖的转换,每一个药用化学家都反应他们希望存在的列表。这就是这个列表的样子。
的愿望清单
1。氟化——交换一个特定的氢氟原子在分子与许多官能团。反应安装difluoromethyl组就好了。
2。杂原子烷基化-一个反应,选择性地高度烷基转移到一个杂原子在有几个戒指,如摘要、氮杂四唑、吡啶酮。
3所示。碳耦合反应一样健壮和多才多艺的传统交叉耦合缝合在一起的脂肪族与控制手性碳原子,理想情况下,。化学家也想要更多的选项类型的前体分子他们可以使用耦合。
4所示。制作和修改杂环化合物——安装官能团的反应——从烷基卤-芳香族和脂肪族杂环化合物,如吡啶,哌啶或异恶唑。反应可以从头开始全新的杂环化合物将是一个奖金。
5。单个原子原子交换反应,可以交换选择性,如交换碳氮原子的戒指。这种化学物质的基因编辑的版本能够彻底改变药物的发现,但从认识可能是最远的。
1。氟化作用无处不在,随时
也许不足为奇的是,一个反应,可以将一个碳氢键转换成氟键的愿望清单。超过20%的所有商业药品氟。2抗抑郁药氟西汀——就是人们所熟知的百忧解——是一个突出的例子。

添加甚至一个氟原子到分子可以增加代谢稳定性和亲油性。反应快速修复放射性氟18到分子也可以对于医疗正电子发射断层扫描。
但是没有直接的方法来替代一个特定的氢原子与氟一旦组装分子结构,解释了Astex的首席科学官大卫•里斯。“如果你问一个药物发现化学家当他们有他们领导分子:“你能把一个氟在每一个职位吗?“总的来说,答案是:“我必须回到一开始的合成和氟化原料开始。”
有不少方法可以使与氟素化合分子。3但是他们都需要安装另一个活性基团——像一个锡或芳烃硼片段,或一个双键或环氧乙烷在脂肪族化合物和交换氟。药物化学家所真正追求的是一个健壮的方式直接交换的H F。
但这些直接氟化反应常常纠结于选择性问题,给regioisomers或over-fluorinating化合物的混合物。由于单一氟几乎不改变分子的反应或物理属性,删除多余的起始物料或后端产品反应并不是微不足道的。
很多同样的东西是真实的反应安装difluoromethyl组。CF2H组bioisostere硫醇或醇——它也有类似的生物属性这些团体,但可以降低药物的毒性和提高其生物利用度。我们越来越多的设计成分子,但化学还没有完全跟上,”说克里斯托弗是不可或缺辉瑞公司资深首席科学家。
为数不多的方法来安装一个CF2H组使用diethylaminosulfur三氟化(DAST),液体变成炸药的uid国际清算银行当加热(diethylamino)硫二氟化物。这是否比使用替代——气态四氟化硫,释放高度腐蚀性氢氟酸在接触水分——是有问题的。有很好的稳固的,你可以拿现成的安装difluoromethyl集团将成为一个伟大的进步,”说,是不可或缺的。
2。当有几个烷基化杂原子
这可能是如此平凡的,大多数人甚至不想思考,但如果它能做这将是非常有用的,”说,是不可或缺的反应他的个人愿望列表的顶部:吡啶酮烷基化。
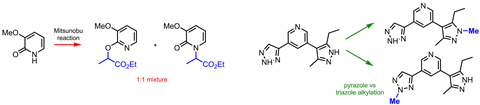
附加任何选择性氮或氧2 -或4-pyridones,到目前为止,不可能的。化学家们通常与野生的混合物N-alkylpyridones alkoxypyridines。这样一个常见和简单的转换,我很难相信还没有一个一般的补救方法与吡啶酮多久出现在药物发现,“我不可或缺说。
尝试找出背后的规则进行了羟基吡啶的双面的反应。4、5但有很多个人因素,包括羟基吡啶的替代模式,烷化剂,用于去质子化,温度甚至溶剂,仍然很难预测或控制,会产生什么反应。
有很多杂环化合物在药物发现库遭受同样的问题。摘要和氮杂四唑例如,分别有两个和三个氮原子。没有试剂组合有选择地修改其中一个——他们太相似的反应。和打消念头想做选择性烷基化的化合物,杂环化合物的有几个。
杂原子烷基化的原因是药物化学家中最希望是这些支架做出好的药物。抗癌药物topotecan和伊立替康功能N烷基化吡啶酮;O烷基化吡啶酮可以在抗疟药。还有一大堆的抗真菌药物,氟康唑,烷基化三唑单元。
3所示。充碳耦合
交叉耦合反应享受巨大的成功因为他们发现在1970年代。反应,加入两个芳香或其他sp2碳原子聚集在一起的帮助下构建有机钯催化剂已成为必不可少的支架。但平面biaryls传统交叉耦合反应非常善于越来越有限,化学家们希望他们的三维耦合。
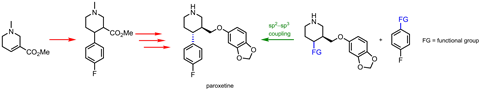
“制药正朝着更高的sp发展3碳含量,有机化学家解释道玛丽沃特森美国特拉华大学的。饱和碳允许您调整第三维度,它可以让你减轻脱靶效应没有戏剧性的变化在其他药物如属性。”一个健壮的反应,碳原子连接芳香族和脂肪族,甚至两个脂肪族片段——是药物化学家所追求的。
“人们喜欢钯的一部分原因是他们知道会发生什么,它的氧化态和氧化还原反应控制,”沃森说。但同时为sp钯的催化循环是完美的2片段,它不与脂肪族耦合伙伴做得很好。不良副反应可以收购,其他时候的反应完全关闭。可能需要的是一个完全不同的金属。
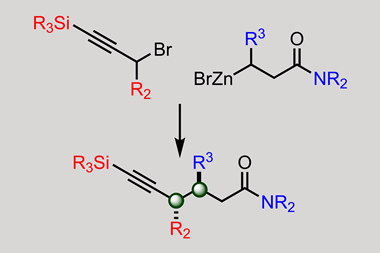
镍,例如,允许不同寻常的转换(如烷基胺之间的交叉耦合和alkylzinc卤化物。6其他金属可以做更多的异国情调的转换。例如,但如何你控制铁和氧化还原活性差异很大?“沃森问道。“什么是正确的配体,正确的条件让它做你想要做什么?”
但无论金属,所有需要pre-functionalisation交叉耦合。将催化剂活性组到耦合伙伴让知道加入碳原子。有时,例如与吡啶,使得这些前兆是一大关键。所以一些化学家想完全摆脱pre-functionalise的必要性。我们的目标是将碳氢键直接转化为碳碳键。
问题是,有机分子都是关于碳氢键。周围有很多人很难告诉一个催化剂。目前,反应依赖于工作区,底物的固有反应性偏见或指导小组,指导催化剂到特定的碳氢键。
即使一个普遍的碳氢键活化反应有一天成为现实,“我认为有时仍将会更容易的预官能团或利用现有的官能团,”沃森说。有机化学的美是我们创建的选项,所以我们可以选择一个最意义为应用程序。
4所示。制作和编辑环
杂环化合物是药物化学家的面包和黄油。大约60%的所有小分子药物有一个杂环核心。目前最常见的哌啶,吡啶和吡咯烷,但也有penams,morphinans和异恶唑25个最频繁的氮杂环化合物。7
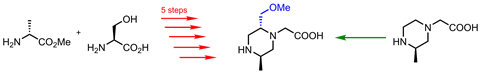
许多小杂环化合物是商用,但不可以成为一个主要的合成头痛。'如果你有multi-substituted吡啶,表面看起来很简单的东西,有时候你意识到为了合成的起始物料可能需要7、8、9的步骤,”说,是不可或缺的。
许多反应为全碳芳烃不翻译杂芳族化合物。氟化试剂能与杂环化合物反应和氧化,而不是使与氟素化合。交叉耦合催化剂往往受到杂环化合物,因为他们坚持永久金属。
有一些方法可以安装官能团芳香杂环化合物在合成,像iridium-catalysed borylation。它安装一个硼组可以被替换为一个取代基的选择。但找出可能发生的反应——混合杂芳族化合物的立体和电子效应发挥作用,是整个论文的主题。8如果你可以先吡啶,廉价,你可以升或其他简单,大量可用的杂环化合物,和你可以绕环和选择性地安装不同的团体,我会的工作,“笑是不可或缺。
当化学家们想要一个脂肪族杂环不寻常的替代模式,它变得更加复杂。通常,这意味着从一个非循环的前兆。无环上的取代基安装分子,然后刻意cyclised在之后的合成。如果cyclisation失败,又回到了起点。
“这伤害我们,因为它是缓慢的,”里斯说。经常的,我们没有时间去做这些化合物所以不要探索这化学空间,即使它可能会成为非常有趣的药物。”
2009年,在英国化学家生物科技公司发表了一篇论文未来的Heteroaromatic戒指。躲在这诗意的标题是一个列表,超过3000个小杂环化合物,尽管他们看起来“综合可行”,从来没有。
十年过去了,一群法国研究人员重新合成的状态通过查看2009年的22例子研究了选为代表。99的小自行车被合成为确切的戒指。六了大支架的一部分。另一个七还没有被描述。
5。原子交换
’对我来说(它)会最有用新的药物发现化学和反应可能与诺贝尔奖获得者合成,”里斯说关于键交换反应。这种转变可能会彻底改变药物化学如果它是可能的。

化学家们不需要担心任何更多关于如何functionalise杂环化合物或如何交叉耦合的几组胺。像一个基因的化学版编辑,反应可以完成分子,目标一个特定的碳原子和换一个氮、氧或硫。
当然这是前所未有的,有些人不喜欢讲这个,因为他们说这是不现实的,”里斯说。但在Crispr-Cas9问世之前,人们不认为目标基因编辑可能。“科学动作非常快,有很多有创造力的人在有机化学中,”里斯说。我肯定会做,这只是一个问题的。”
的Baeyer-Villinger氧化和贝克曼重排是最接近的两个反应一个假想原子交换。都是100多年前发现的,都是简单:他们插入一个氧或氮循环酮。但都添加一个原子而不是取代。
确认
谢谢你从麻省理工学院的康斯坦丝诺依曼,和本·Glasspoole Kaelyn Wilke从微孔σ和肯尼斯•Schwieter有益的讨论。
引用
1 D G布朗和J博斯特罗姆j .地中海,化学。,2016,59,4443 (DOI:10.1021 / acs.jmedchem.5b01409)
2 G K萨普拉卡什和F王今天的化学,2012,30.,30
3 C N诺伊曼和T里特,Angew。化学。Int。。,2015,54,3216 (DOI:10.1002 / anie.201410288)
4 M Breugst和H·迈尔,j。化学。Soc。,2010,132年,15380 (DOI:10.1021 / ja106962u)
5 M C Torhan N P皮特和J D威廉姆斯,四面体。,2013,54,3926 (DOI:10.1016 / j.tetlet.2013.05.054)
6 S冷藏室等,j。化学。Soc。,2019,DOI:10.1021 / jacs.9b00111
7 E Vitaku D T T Njardarson史密斯和J,j .地中海,化学。,2014,57,10257 (DOI:10.1021 / jm501100b)
8 M拉森和J F Hartwig,j。化学。Soc。,2014,136年,4287 (DOI:10.1021 / ja412563e)
9 K Passador Thorimbert和C Botuha,合成,2019,51,384 (DOI:10.1055 / s-0 037 - 1611279)
进一步的阅读
D C布莱克莫尔等,Nat,化学。,2018,10,383 (DOI:10.1038 / s41557 - 018 - 0021 - z)
J博斯特罗姆等,Nat。启药物。,2018,17,709 (DOI:10.1038 / nrd.2018.217)
K R坎波斯等,科学,2019,363年eaat0805 (DOI:10.1126 / science.aat0805)
T Cernak等,化学。Soc。牧师。,2016,45,546 (DOI:10.1039 / C5CS00628G)







1读者的评论