



开发治疗罕见疾病的药物并不总是经济可行。克莱尔桑塞姆看着最近的一些成功的故事
商业药物发现异常昂贵,成本的一个分子从基础研究到临床应用的估计超过十亿美元。如果一个公司要收回这个金融投资而收费,卫生系统是准备付出代价的,它需要出售大量的药物开发。
该行业的理想是慢性疾病,意味着病人呆在药物多年。理想情况下它还将主要影响人们在富裕国家和有一个大的患者人群。病人被诊断患有罕见疾病因此产生显著的缺点。这并不完全是一个经济问题:如果只有少数-甚至几千病人拜访,建立临床试验提供足够的统计能力检测效果也变得极其困难。
越来越多的人受到比似乎直觉可能罕见疾病。在欧盟,估计有30 - 40几百万人患有一种疾病列为罕见。这种差异来自大量的这种疾病。“罕见病”的定义司法管辖区之间的不同:美国食品和药物管理局(FDA)只考虑患病率(它必须影响不到200000个人在那个国家)而欧盟只包括疾病危及生命或慢性衰弱。无论使用哪种定义,术语包含超过5000个不同的条件。大约80%的这些遗传或主要遗传原因,和许多在婴儿或儿童早期诊断。
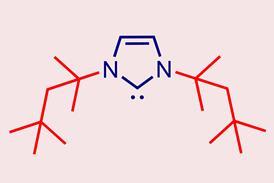
一些国家已经颁布了所谓的孤儿药立法,提供科学建议,降低费用和提高药品专利保护公司发展中这些条件。欧洲药品局(EMA)和FDA采取类似的方法,紧密合作。我们指定一个药用产品作为其赞助商孤儿如果可以证明它是用于诊断、预防或治疗一种罕见的疾病或一个激励药物开发不足,这给病人提供了一个很大的好处,”说乔迪Llinares加西亚产品开发和科学支持主管教育津贴。赞助商通常,但并非总是如此,公司:“个人偶尔申请孤儿药物名称和取得成功,”添加了克里斯蒂娜拉尔森EMA的孤儿药物。
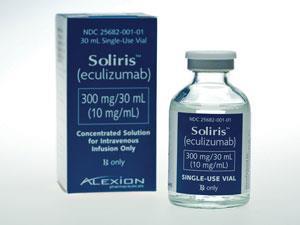
这项法案使药物开发的一些非常罕见的疾病。Soliris (eculizumab)是一种单克隆抗体许可两件疾病的治疗包括血液凝结,阵发性夜间血红蛋白尿和非典型溶血性尿毒综合症。这种药物——可能在英国大约有200患者受益——仍然是有争议的,因为它的高成本,估计约400000美元(£260000)每年每个病人。这使得世界上最昂贵的药物之一。
常见的罕见的疾病
囊性纤维化,从而影响一个2000 - 4000年白种人新生儿,是最常见的一种罕见的疾病。大约有10000人感染了这种疾病在英国。这是一个经典的孟德尔遗传隐性模式。这意味着那些继承了一个错误的复制基因的囊性纤维化跨膜电导受体(雌性生殖道)将是健康的,但是能够把疾病传染给他们的孩子,而那些继承了父母双方的错误的副本将有疾病。CFTR蛋白的检测基因形成一个通道,通过它,氯离子通过细胞膜的细胞。这些通道是重要的生产粘液,汗水和消化液。囊性纤维化患者,这种蛋白质的缺乏工作副本,遭受一群肺癌和消化问题。
不是所有的雌性生殖道突变,因此,但是,并不是所有雌性生殖道的病人,都是平等的。大约一半的囊性纤维化患者在英国有两个拷贝一个最常见的基因突变,哪一个苯丙氨酸残留是失踪。这产生一个不稳定的和完全不活跃的蛋白质,和严重的疾病。病人的雌性生殖道蛋白质包含单个突变氨基酸,相比之下,可能会产生一个稳定的蛋白质功能效率低下;在英国最常见的是所谓的凯尔特突变,G551D。带有这种突变的蛋白质在细胞表面形成一个通道,形式是正常的,但很少打开。
直到最近,才可以治疗囊性纤维化的症状。第一个目标药物潜在的缺陷,Kalydeco (ivacaftor)是由顶点药品(总部设在波士顿,美国),并于2012年授权。这是一个相当小的和简单的分子——dihydroquinoline酰胺——结合雌性生殖道的孔隙通道,改变其闸门机制经常打开通道。它可以,不过,只把有缺陷的突变患者像G551D蛋白质仍表达在细胞表面。
简·戴维斯在儿科呼吸医学顾问在伦敦帝国理工学院,英国,参与临床试验的药物的患者在6岁和至少一个G551D等位基因。我们显示药物相当成功地改善患者的肺功能和在维护改进好几年了,”她说。但是,争论其成本,每人每年约300000美元在美国(英国成本尚未公布)。
Kalydeco的发展是一个重大突破在囊性纤维化,但只希望帮助少数患有这种疾病。顶点目前正在调查Kalydeco的组合lumacaftor在CFTR突变导致病人很少甚至没有检测蛋白质在细胞表面,并积极迹象的III期试验。基因疗法修复或替换有缺陷的基因也在调查中,英国囊性纤维化基因疗法财团最近完成了一个小二期临床试验正常的囊性纤维化基因交付使用脂质体载体进入细胞。数据还在分析。
男孩的网络
杜氏肌营养不良症(DMD)囊性纤维化的模式类似,戏剧性的进展是少数患者治疗疾病。DMD只发生在男性,影响约1 3500年新生男婴。男孩DMD继承一个有缺陷的复制抗肌萎缩蛋白基因编码的一种蛋白质,这种蛋白质的复杂连接肌肉纤维的一部分周围的细胞外基质。他们通常是坐在轮椅上的青少年,很少活到三十几岁。
肌营养不良蛋白的基因编码是我们最大的基因组-超过250万个碱基对,产生一种拥有超过3500个氨基酸的蛋白质。这个基因,像大多数在人类基因组中,包括许多的DNA代码的部分蛋白质-外显子-穿插非编码基因内区。许多致病突变在肌营养不良蛋白涉及一个或多个外显子的缺失,这一过程会产生两种不同的疾病。最外显子缺失导致催化作用的蛋白质的分子机器翻译,核糖体,阻止其在信使RNA所以没有蛋白质的形成。在少数情况下,然而,核糖体能够跳过被删除的外显子,继续翻译剩余的蛋白质,因此产生一个短的蛋白质,保留了一些功能。男孩与这些突变温和得多肌肉营养条件,贝克尔肌肉萎缩症,能活到中年甚至老年人群中——尽管总是有些残疾。
Sarepta疗法位于剑桥的我们,是几家公司发展中方法生产缩短但是功能强大的肌营养不良蛋白蛋白之前,患者由于基因突变,产生了没有。技术,称为外显子跳过,删除一段信使rna基因突变后,使核糖体继续翻译从一开始的下一个外显子,而不是停止。这产生一个短,部分功能的蛋白质,本质上改变DMD的温和的疾病,可能会像贝克肌营养不良。
公司开发phosphorodiamidate吗啉代寡聚物(pmo),它包含DNA碱基连接到一个骨干组成的六元吗啉环与phosphorodiamidates五元的(而不是自然核酸核糖环磷酸二酯连接的链接)。交付给一个细胞,PMOs绑定到靶向RNA和屏蔽RNA的细胞机制。这有助于修复有缺陷的RNA和恢复翻译所需的蛋白质。

公司的主要产品是eteplirsen PMO束缚专门51外显子序列的抗肌萎缩蛋白mRNA。二期临床试验已经表明,这种药物可以恢复肌营养不良蛋白的合成,防止一些肌肉在这种安全恶化和年轻十几岁的男孩仍然能够走路。Eteplirsen可以治疗大约13%的男孩demand media,比例高于目标可以通过药物治疗其他外显子,“Sarepta首席执行官克里斯Garabedian说。”然而,开发类似的药物基于相同的方法将允许我们目标其他突变和地址DMD人口的80%。
这种方法的缺点是,eteplirsen——比如Kalydeco囊性纤维化,只有少数患者有效。另一种方法可能治疗每一个DMD病人刺激另一种蛋白质的生产,拉到,有一个类似的功能,但由未出生婴儿自然合成。
凯戴维斯医学研究委员会的功能基因组学单元在牛津大学,英国史蒂夫·戴维斯牛津大学化学系的这种方法。“史蒂夫和我开始峰会制药在2003年我们的多年的研究转化为肌营养不良蛋白和DMD诊所,”戴维斯说。欧洲企业的激励措施较少开发新药比现在对于罕见疾病,最好的出路似乎开始我们自己的。”
峰会的研究人员使用高通量筛选选择化合物诱导拉到和他们的生产铅分子,SMTC1100,目前在临床试验中。我们不知道这种化合物的确切的作用机制,但是我们知道它在转录水平工作:也就是说,它引发的生产拉到mRNA的蛋白质,”戴维斯说。
SMTC1100完成了第一阶段的研究没有严重的毒性问题,现在的Ib期临床试验与DMD少量的男孩。一些男孩服用此药显示临床改善但团队一直受到病人的反应各不相同。看来,一个男孩的饮食影响药物吸收的方式和研究正在关注是否改变药物管理的方式,目前口头传递,会改善其吸收。
团体意识
尽管孤儿药法案提供的激励,企业仍不愿投资于罕见疾病的早期药物发现项目。SMTC1100突出的发展公司和学术团体之间的合作的方式可以帮助填补这一缺口。有一个第三方,可以几乎同等重要:患者和他们的家庭。一些疾病的社区拥有自己的国家组织将病人一起来支持研究;囊性纤维化患者和肌肉萎缩症是由这些。然而,一些疾病是如此罕见,病人社区太小,形成有效的支持团体。在这里,保护伞组织,如病因——“罕见疾病患者在欧洲的声音”,全球基因总部设在美国,但活跃在世界范围内,可以有帮助。

全球基因与一个独特的病人的组织,PatientsLikeMe公司一百万年,建立一个网络病人患有罕见疾病。PatientsLikeMe公司成立于2004年,由兄弟詹姆斯和本杰明·海伍德和一个家庭的朋友杰夫•科尔支持第三个海伍德哥哥,斯蒂芬,他开发了罕见的疾病肌萎缩性脊髓侧索硬化症(ALS)。他们的目标是建立一个在线网络通知病人症状和治疗方法和讨论他们的资源池。今天,有超过300000名患者注册为用户。”代表我们有超过2000种不同的疾病,从常见的乳腺癌和帕金森病等一些只有少数家族从未发现,”保罗·威克斯说创新的副总裁PatientsLikeMe。
这些网络的通知,联系病人开始影响临床试验,威克斯解释道。2007年一群-巧合ALS患者发现一份报告的一个小试验表明,碳酸锂用于治疗双相情感障碍,可以控制他们的症状。几个月后,160名患者来自许多国家一起获得药品标示外,通过PatientsLikeMe监控自己的症状和提供统计证明,锂是无效的:四大官方试验证明了病人,而不是科学家们进行了第一次试验,是正确的。”
基因组测序
有一些病人没有治愈的希望,甚至参与临床试验,因为他们缺乏诊断。但有一个几乎可以肯定的方式跟踪甚至最模糊的疾病:通过病人的完整基因组测序。测序成本已经下降到一个点时变得可行。的目标之一100000人基因组计划由基因组学英格兰在2014年推出,是完整的基因组序列成千上万的罕见疾病患者和他们的近亲。这是一个通用的方法,可以应用于诊断任何罕见的遗传疾病,就可以通过NHS在任何地区,”说珍妮特·桑顿欧洲生物信息学研究所(EBI)主任Hinxton工作组的主席,设置项目。
罕见疾病患者的基因组定序很可能偶尔会接缺陷基因也被牵连到更常见疾病,,如果病人是幸运的,药物已经许可用于治疗常见的疾病将帮助罕见。这种协同作用反过来也可以工作。的治疗目标中心的验证(东森)已经建立在EBI Hinxton——共同,邻近的桑格研究所(其中三分之一的原始人类基因组测序)和制药巨头葛兰素史克公司——选择,优先考虑和研究各种疾病最好的药物靶点。有时学习一种罕见的疾病的遗传原因将提供洞察适当的一个共同的目标,multigenic同样的途径,”伊恩·邓纳姆说,科学主任东森。例如,一些NOD2基因突变导致罕见的炎性疾病,布劳综合症,而其他变体与炎性肠道疾病的风险增加有关。”
这些例子显示患者的前景至少有几个已知的成千上万的罕见疾病正在转变,由于开明的立法,基因组学和联合技术,并非最不重要的是,病人自己的参与。有另一种类型的疾病,然而,在药物开发的金融挑战,而相似但更光明的前景。这些被忽视的疾病,如血吸虫病和恰加斯病,影响数以百万计的人来说,几乎所有的人都是穷人。
克莱尔桑塞姆是一个基于科学作家在伦敦,英国








还没有评论