
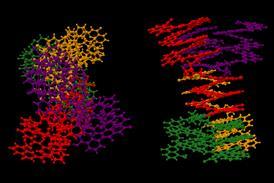



第一次,化学家已经使用量子力学计算预测的几种可能retrosynthesis路线。这种计算方法允许他们缩短的总合成六环萜来自27个步骤9。
依靠文学先例或人类直觉设计总合成时可能会非常棘手。很难预测与多个官能团的化合物或不寻常的空间属性的反应。不幸的化学家们可能要花几个月的时间整理一个天然产物反应一次只发现一个步长序列结束时不起作用。这通常意味着从头开始整个合成。
在预印本刊登的一篇研究论文ChemRxiv,蒂莫西·纽豪斯和他的团队来自耶鲁大学,我们表明,密度泛函理论(DFT)造型的猜测,总合成。
DFT计算模型原子之间的相互作用和估计不同化合物的行为反应。但造型通常做回想起来,例如研究反应机制——纽豪斯决定使用它的预测能力。
棘手的萜烯
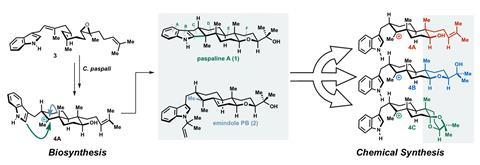
团队解决两种萜烯合成:paspaline A和emindole PB。这些真菌萜烯干扰信号通路在某些癌细胞。前者的合成之前采取了27个步骤——团队在9——而后者从来没有。
Paspaline结构人工催的吲哚环戊烷环融合。以前,化学家们建造了吲哚环在最后几个步骤对总量的合成。灵感来自于paspaline的生物合成,纽豪斯和他的同事们决定把现成的吲哚的类固醇。一个棘手的cyclisation最后的合成将完成最后的类固醇环——一个冒险的举动,说博士研究员和第一作者Daria金。
选择最佳的三个可能的前兆至关重要的转变,团队进行DFT计算取代基分子的远端如何影响反应结果。与二环缩酮化合物取代基变成了最有利的能量——这是一个有点令人惊讶的结果,因为它是比其他选项sterically紧张。
(DFT计算)允许我们评估基板的方式修改不明显改变转换的结果,”纽豪斯解释说。我们修改的功能,可以从反应非常远端网站,似乎并不会有这样的影响的关键一步——但它确实。
DFT做了另一个不寻常的预测被证明是正确的:化合物会形成一个反式融合环系统,虽然反应底物导致报道相似独联体融合戒指。
溶剂在干草堆
然而,DFT无法预测哪种溶剂最适合或温度运行反应。每个基板我们看着就像一个干草堆,和在一个干草堆针最佳反应条件,“金解释说。提取的过程中,针仍非常困难和费时,但是DFT与你有更多的保证你的干草堆包含针。”
此外,金解释说,DFT只能如果用户知道反应的机制。的计算只是一样好你的信息。DFT不会识别机制。”
[方法]将允许多重假设检验并帮助优先路线可能是最可行的,“博士后研究人员说卡洛琳Ladd,在天然产物的合成莎拉·赖斯曼在加州理工学院的,我们。因为许多化学家们已经在使用DFT软件,它应该简单适应retrosynthesis计划,她补充道。这是一个伟大的方式来利用已经成熟的计算方法。
“这会是有趣的,看看计算计划筛选潜在的基板是将在更广泛的社区流行的东西,”金说。纽豪斯认为,DFT计算甚至可以增加合成的预测能力规划等工具Chematica。
引用
J D E Kim E茨威格和T R纽豪斯,ChemRxiv,2018,DOI:10.26434 / chemrxiv.7322330.v1





还没有评论