
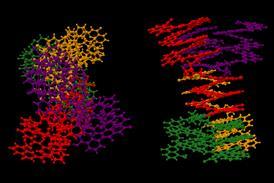



快速、可靠的新分子动态模拟方法将加快研究由葛兰素史克在调查中
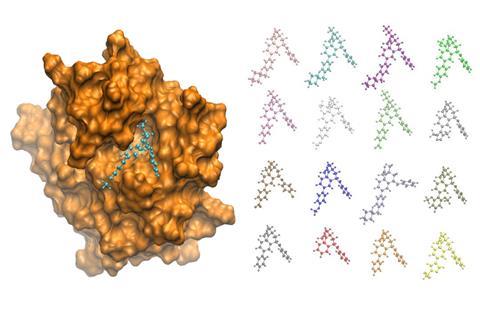
英国科学家设计了两种分子动力学模拟方法,他们说可以为基因药物开发。1制药巨头葛兰素史克(GSK)已经使用它们来模拟化合物与蛋白质交互部分称为bromodomain,与癌症和炎症性疾病有关。葛兰素史克的结论”,需要进一步调查的方法,因为它有可能大大减少发现和开发时间的,说Peter Coveney来自伦敦大学学院(UCL)。
制药行业定期使用分子动力学模拟蛋白质之间的相互作用和药物的候选人。然而,伦敦大学学院的研究小组最近发现一个根本性的随机性使得单一的模拟unreproducible。然而大多数出版物报道一个模拟的行为,“Coveney说。
Coveney参与€2.1亿(£1.8亿)欧洲人虚拟生理人的倡议支持个性化医学。所有这一切的关键是确保一个可以产生快速、准确、精确和可再生的预测,”他说。可以帮助排药物对精确的每个人的基因编码的蛋白质结构。
因此他的团队设计了一个新的分子动力学方法计算结合亲和力-化合物时释放的能量槽成蛋白质基于热力学集成(TI)的方法。因为它很难计算出绝对的约束力的亲和力,TI认为差异绑定两个有机化合物绑定到一个蛋白质的亲和性。新方法模拟逐渐转换从一个化合物,采取几个模拟快照后几个小步骤的“炼金术”过渡。2
平均计算,包括在每一个中间点的变换,给出化合物的相对亲和力为一对,Coveney解释道。这种方法比以前的更可靠和更少的受到随机性方法使用一个转换步骤。计算的速度也很快,因为算法设计,这样他们可以并行运行,独立,适合现代超级计算机。
团队的第二种方法计算绝对约束力的亲和力,也使用大规模的平均。由于这是一个大的挑战的方法不太准确,但可以避免结构性限制之间转换时面临类似的化合物。
科林Sambrook史密斯计算科学和信息学主任Sygnature发现在诺丁汉,英国称bromodomain结果“令人鼓舞”。其他蛋白质的作者提到扩大这项工作,这将是等待感兴趣,”他说。他补充说,早期的一个关键挑战是构建软件来处理大型数据集生成的方法。
引用
1 S广域网等,j .化学。理论第一版。,2016,DOI:10.1021 / acs.jctc.6b00794
2 P Bhati等,j .化学。理论第一版。,2017,13,210 (DOI:10.1021 / acs.jctc.6b00979)





还没有评论