


计算研究使用量子计算可以确定最佳催化剂提供了一个框架
美国研究人员已经开发出一种新的战略预测双金属催化剂。他们已经把它好好利用识别nickel-platinum催化剂,他们声称是最好的氨分解反应-一个潜在的重要反应如果氨成为储存氢的一种重要手段。
对催化剂进行预测,包含多个金属是复杂的,因为一个混合的属性不一定是父母中间的金属。然而,这仍然文学中的常见的假设,说Dionisios Vlachos,这项新研究的作者之一特拉华大学。他的团队开发了一个解决这个问题的方法,利用密度泛函理论(DFT)计算,基于量子力学的计算。
密度泛函理论的计算做的是他们占催化剂的结构,“Vlachos解释道。'你需要考虑原子的独特的建筑空间中,他们实际居住,为了能够预测正确材料的属性。以前使用的简化方法,他说,不占的化学反应。
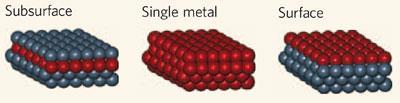
Vlachos的团队利用DFT计算筛选图书馆microkinetic模型——氨(NH详细的反应机制3)分解,与不同的双金属材料作为催化剂。指出单一金属催化剂,氮在催化剂表面的结合能是催化活性的一个重要指标,他们为混合金属计算这个值。1 nickel-platinum催化剂的氮结合能接近当前的最佳单一金属催化剂,钌。实验和研究人员预测出生,活动从50吗?C,比350年?C钌。
工作的真正价值,然而,在创建一个框架,用于识别双金属催化剂一般Vlachos说。铂钌可能不是最好的替代品,因为它的成本,但Vlachos相信同样的原理可以应用于寻找更便宜的材料,和其他双金属催化剂的反应,包括一氧化碳,氢气的选择性氧化净化和碳氢化合物的加氢。
催化专家老人Hviid克里斯腾森为催化工作公司Haldor Topsoe在丹麦,有兴趣看到越来越多组向下计算路线。”这是一个非常有效的方法来缩小可能的巨大范围催化剂的候选人,”他说。”,这个反应本身就是有趣,因为氨是一种无碳能源载体。
海莉桦木
引用
et alNature化学,2010年,DOI: 10.1038 / NCHEM.626





还没有评论