


基于新分子的方法更容易评估反应计算的精度
瑞士的科学家们发明了一种方法来警告化学家当他们计算分子体系的反应应该谨慎对待。1真正的预测,我们需要知道理论方法产量结果影响大的错误,”说马库斯苍鹭”从苏黎世联邦理工学院,瑞士联邦理工学院。“这就是我们的方法的能力。的使用方法,苍鹭”和他的博士生格雷戈尔Simm高度误差密度泛函理论(DFT)计算整个催化nitrogen-to-ammonia转换周期。
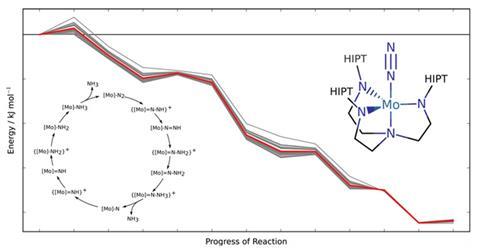
计算确定反应的势能,大多数化学家使用DFT,快速,但往往不准确。误差范围基于DFT的评估结论很难得到,苍鹭”解释道。我们需要在可见误差,我们计算在一个特定的方法,而不是从比较最大的参考数据准确性,”他说。这种准确的数据通常是不可用的。
苍鹭”强调,其他研究人员已经开发了能量密度泛函近似DFT的核心,为固体物理学产生误差。2然而,这种方法不转移实际上从基准化学系统的科学家研究用DFT。缺乏可转让性是特别有问题的兴趣苍鹭”和Simm的过渡金属配合物。
在他们的新方法中,两人做了一个统计上的两套信息之间的联系。第一是DFT估计的比较和参考数据。第二个比较最好的结果从一个密度泛函优化的结果剩下的泛函,这类产生置信区间。连接这两个数据集产生散射与真实错误当参考测量并不是可用的。
苏黎世团队优化他们的泛函,参数是特别适用于分子与金属中心。虽然这种限制的适用性re-optimised泛函,,它增加了可转移性相似的分子和允许有意义的错误评估,苍鹭”说。虽然这意味着所有化学系统需要集中参数化,他们正在试图开发一种更一般的方法。
阿伦·科恩工作在提高DFT泛函,剑桥大学的英国,指出常用的泛函,还有大型的系统误差,尤其是在催化。他所说的新方法“有趣”,但强调固有的权衡牺牲具有普遍适用性。人申请额外的努力但回报你增加信心的结果,”科恩说。
引用
1Simm G N和M苍鹭”,j .化学。理论第一版。,2016,DOI:10.1021 / acs.jctc.6b00318
2J J·莫特森等,理论物理。启。,2005,DOI:10.1103 / PhysRevLett.95.216401





还没有评论