




一种新的神经网络可以帮助化学家计算吸附能量有效且高效地——一百万倍的速度比最先进的方法。再一次,人工智能已经被证明是一个强大的工具来加速发现,特别是在多相催化领域。催化剂的改进设计和开发可以创造更清洁的化学过程,并使探索创新的生物量和塑料等原材料。
”(我们的神经网络)的催化剂多相催化,”说Nuria洛佩兹从ICIQ塔拉戈纳,西班牙,该研究。传统上,计算化学家计算吸附能量和密度泛函理论(DFT),这有几个局限性。DFT是非常耗费时间,加上并不适合非常大的分子,”她补充道。的新的神经网络算法预测大分子在金属表面的吸附能数量级更快,”洛佩兹说。在多相催化、活动实际上是与吸附能量有关。最终,准确估算吸附的能量可能导致更有效的实验在实验室里。
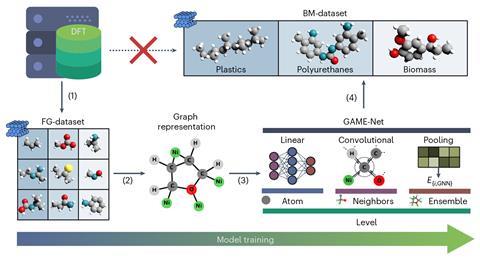
其中一个技巧是将分子表示为数学“图”——结构与几个节点相互连接的链接。在这个简单的系统中,节点代表不同元素的原子,和链接说明化学键。这是一个自然的代表分子,就像使用balls-and-sticks模型,”洛佩兹说。这个概念已经采用分子模拟,但仍相对未知的多相催化,因为很难简化结构表面的。研究人员解决这一挑战通过创建图表的催化表面,选择较小的原子与分子的数量。
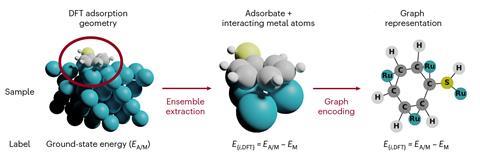
团队训练算法,称为GAME-Net,小分子,具有基本功能组胺、酰胺、酯类和芳烃,等等。模拟的表面,不同方面的数据集包括14金属框架。我们已与DFT计算了小分子的吸附能量滋养神经网络,”洛佩兹补充道。的算法迅速学习化学的基本概念,并成为能力计算大分子的吸附能以最小的错误,”她解释说。
新的神经网络是一个激动人心的工具在世界上的多相催化的,说侬Artrith在机器学习和计算化学专家在荷兰乌得勒支大学的。它克服了长期挑战的造型大分子之间的相互作用与催化剂的表面…和预测吸附能量以闪电般的速度,”她补充道。新的神经网络可以推动生物质转化的催化剂的发现,导致可持续生产的化学物质来自可再生资源。“DFT相比,模型的速度和准确度让人印象深刻,“Artrith补充道。”这一创新铺平了道路…奔向一个更干净、更绿色的未来。”
虽然“吸附能量通常与催化活性,进一步措施可以在实验室里测试的趋势与预测神经网络的比较,Artrith评论。洛佩兹说,准确的实验测量与传统的热化学吸附能量技术需要大量的时间,和一些实验数据库可供比较的目的。然而,团队希望促进伙伴关系探索实验在不久的将来努力。
此外,研究人员将很快开放工具化学社区,创建一个用户友好的网站,使用简单的输入等结构,微笑字符串,PubChem数字和分子的名字。然后,该网站迅速模拟所选金属表面的吸附能。的开放和合作这个项目的核心,一个专家共同努力在加拿大,瑞士和西班牙,”洛佩兹说。通常,DFT,单个模拟大分子的吸附能将天在一台超级计算机,”她补充道。由于我们GAME-Net神经网络,现在你所需要的是笔记本电脑。
引用
年代Pablo-Garcia等,Nat。第一版。科学。,2023,DOI:10.1038 / s43588 - 023 - 00437 - y






还没有评论