


研究显示,以及连续溶剂方法失败solvent-phase反应机制的计算研究,提供一个替代溶剂建模策略
量子计算模型可以捕捉在复杂solvent-phase物理化学反应机制,新研究表明,尽管最近声称相反。1这项新研究引入了一个静态的量子化学计划以显式溶剂造型。它可以作为一个替代方法来看看液相反应机制在分子动力学模拟是不可能的。
这个项目是发起的出版,而挑衅的研究2德州农工大学的丹尼尔Singleton,解释道约翰·基思美国匹兹堡大学的领导工作。单例的研究复杂Morita-Baylis-Hillman反应机制;烯烃和醛反应耦合催化了叔胺。它突出了使用连续溶剂模型的负面影响,频繁使用的量子化学计算液相溶剂的反应机理的研究。除了钉下一个完整的反应机理,外卖的纸是所有标准量子化学模拟程序是错误的,”基思解释道。
自然,这个结论中创建一个热点造型社区。虽然有些觉得单例的结论过于广泛的负面,基思的团队看到他们作为一个丰富的学习工具。我们没有不同意任何结论由单例,但我们很惊讶很差的计算模型使用连续溶剂方法执行,”他说。
基斯认为,从根本上少了什么。也许solvent-reactant交互是重要的机械的途径?如果是这样的话,这将是更准确,包括明确的溶剂分子,而不是使用一个连续的模型。验证这个假设并不容易:“我首先想到的是6 - 12个月最终是更具有挑战性的工作。幸运的是,我有一个很有才华的1年的博士生,Yasemin Basdogan保持专注和永不放弃的项目——或者我!”基斯说。
他们最初的计划是使用复杂的方法给精度高于溶剂化作用能量的单例的研究。但是结果并没有增加。它还得知一个不同的研究小组,由杰里米·哈维在比利时和KU鲁汶Raghavan Sunoj的印度理工学院孟买,有类似的想法。哈维和Sunoj的研究3报道溶解能量同意与单例的结果,但他们使用分子动力学方法,耗时和技术要求。
没有更多的猜测
基思改变了他最初的焦点假说:从高层理论转向理解标准的建模方法失败的原因。他表明,连续溶剂模型不能描述当地的溶解效果很好。这可能导致机械的步骤如质子穿梭和电荷转移是模仿不佳。作为替代,他开发了一种策略,可以由任何一个有量子化学的一般理解。
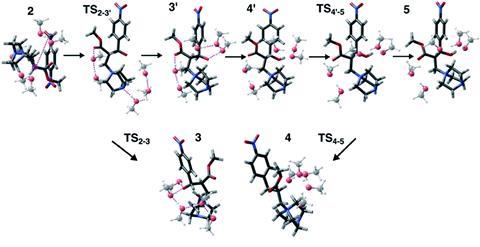
战略的第一步是确定假设的反应物状态与不同数量的溶剂分子周围聚集,然后在全球范围内优化这些。只有少量的溶剂分子需要考虑:基斯的团队发现,使用五个溶剂分子是足够的例子。接下来,您需要使用字符串增长方法系统地探索反应途径的计算消除模型与现实障碍反应步骤发生的实验。日益增长的字符串方法是一个简单的反应路径和过渡态发现工具。最后,您应该能够确定一个模型与一个合理的反应概要文件使用能量从可信程度的计算或实验数据是否可用。一个可选的额外的步骤是确定亚稳中间状态之间的势垒高度为一个更完整的图片,使用越来越多的字符串的方法。
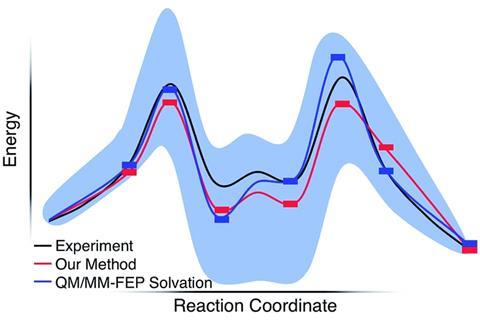
由此产生的溶剂化作用能量同意的结果单和Sunoj尽管只有明确造型少量的溶剂分子。保罗·齐默尔曼,从密歇根大学理论化学家,我们开发了自己的代码字符串增长方法计算。他说基斯的工作把睡过去的指控“量子化学模型可能不只是nonquantitative,但即使定性误导的。这项研究表明,“身体正确的模型可以以合理的成本,没有“猜测”,和化学模拟可以给正确的答案是完全正确的。”
基斯指出,“任何人都使用这种方法应该意识到这不是一个替代的完整和可靠的模拟,计算动态社区开发了多年。这个方法只会提供一个能源配置一个配置在一个途径。“用户需要做一些试验和错误尝试找到一个合适的配置,同意化学直觉和可用的实验数据。
引用
1。Basdogan Y和J基思,化学。科学。,2018,DOI:10.1039 / c8sc01424h(本文是开放访问。)
2。R E Plata和D单,j。化学。Soc。,2015,137年,3811 (DOI:10.1021 / ja5111392)
3所示。Z刘等,理论物理。化学。化学。理论物理。,2017,19,30647 (DOI:10.1039 / c7cp06508f)(本文是免费访问到2018年7月30日。)





还没有评论