


需要使用波函数或密度泛函理论(DFT)计算来确定电子密度绕过了机器学习模型。它将使化学家快速确定属性取决于电子密度的大型系统,如范德华力、卤键和C-H-π交互。这些非共价相互作用能洞察绑定的主-客体系统或支持对映体在反应途径,中间体和过渡状态可以稳定微妙的景点。
电子密度分布是最有力的工具之一的处理计算化学家。从电子密度、属性等费用、偶极子和静电相互作用能量可以确定。准确占这些至关重要的预测能力等量子化学方法计算红外强度或确定非共价相互作用。
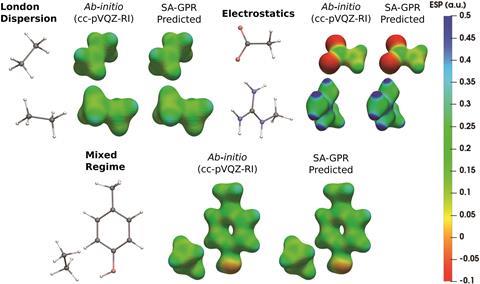
计算的电子密度可以挑战和耗时的大型系统使用传统的波函数或DFT方法。为了克服这个问题,克Corminboeuf,米歇尔Ceriotti和他的同事们在瑞士联邦理工学院(EPFL)已经开发出一种机器学习模型,该模型可以预测的电子密度只有原子坐标。的突破是能够准确地预测,在几分钟内最多复杂分子的电子密度量子化学计算,没有任何的团队成员阿尔贝托·法布里奇奥解释道。
我认为这是一个很有趣的方法,无论是预测错误和可转让性小型和大型系统,”评论娜塔莉Fey研究计算无机化学布里斯托尔大学的英国。
量子技巧更好的机器学习
机器学习模型依赖于一个巨大的训练集的小分子二聚体。这些二聚体有其电子密度表示为基础集,类似在正常计算化学计算。增加预测的准确性的诀窍电子密度是使用辅助基础设置为resolution-of-the-identity方法——近似设计,有助于加快计算积分的两个电子。的关键发展工作,与先前的研究相比,现在研究人员介绍了辅助函数来有效地parametrise密度预测,”言论马库斯苍鹭”理论化学家在苏黎世瑞士联邦理工学院。
“标准基础集构造尽可能近似波函数,而辅助基础集内resolution-of-the-identity近似被设计用来模拟单电子密度,“Corminboeuf解释道。通过使用这些辅助基础设置,下面的错误预测密度的减少0.5%,比> 10%的错误更准确更常用cc-pVDZ等基础设置。
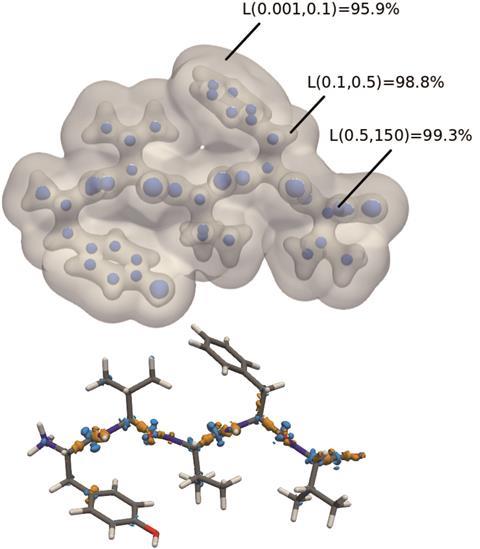
一旦机器学习代码训练密度泛函计算的电子密度小的二聚体,能够准确地预测电子密度的化学性质不同,和更大的系统探索。77原子的电子密度多肽脑啡肽对DFT计算成功地预测在1.4%,在一系列的八大多肽,平均误差仅为1.5%。这个电子密度比标准的DFT方法实现更快,更快的获得至关重要的电子密度和进一步洞察非共价相互作用在这些大的多肽。
然而,Fey指出,当前模型的“近视”仍是一个问题。这个指的是机器学习模型代表分子4大atom-centred环境。因此,远程交互并不完全。
Corminboeuf表明一种可能的解决方案指出“可以解决这个问题通过修改我们代表我们当地的化学环境的方式。非本地信息进行编码,可以制定框架来表示环境潜在的形式,综合了所有空间。”现在,新开发的机器学习模型将允许大量的快速预测可能性只要原子坐标,是否来自x射线晶体学经典分子动力学模拟。






还没有评论