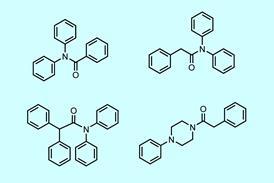


Decarboxylative交叉耦合产生苄基氟化物
有时一块新的化学完全填补了缺口。虽然可能不包括小说反应的一项研究中,它仍然可以是非常重要的如果它优化条件,一套新的基质,如那些在生命科学中使用。这些基质常常缺少的“改变游戏规则”的报告,描述新的反应,所以它往往是后来出版物,巩固反应的有效性。
在过去的10年中,photoredox催化无疑是最具创新性的合成化学领域。其中最重要的反应之一是decarboxylative交叉耦合的羧酸和芳基卤化物。通常,一个铱催化剂和可见光驱动脱羧生成一个烃基困,再加上(杂)芳基卤化物的镍催化剂。

有些令人失望的从药物化学家的角度,交叉耦合反应α-fluorocarboxylates一直难以捉摸。未来的苄氟化物是可取的氟能稳定氧化脆弱的代谢酶的活性,以及给分子构象刚度通过偏转效应或通过调整其物理化学性质。鉴于原料也很容易准备,很高兴看到领导的一个团队明Joo Koh新加坡国立大学找到一种方法这个反应工作。1
团队开始考虑使用的挑战α-fluorinated酸在反应中,与α-fluorophenylacetic酸给“标准”条件下只有15%的收益率。目前还不清楚如果任何酸仍反应后,虽然类似的可比氧化电位建议激进的一代不是至关重要的。
假设烷基容易形式,团队然后使用密度泛函理论(DFT)提出一个机械化的途径。直接氧化加成的芳基卤化物倪(0)是打折的,因为它将涉及高能过渡状态,留下两个更加积极合理的途径:激进倪(0),紧随其后的是氧化加成的芳基溴化和随后的激进分裂给一个正方形平面Ni (二世)的物种;或附近barrierless亲核芳香替换(SNAr)之外的芳基溴化和后续添加生成相同的复杂。异构化四面体倪(二世)物种和添加F-alkyl激进然后允许内在领域债券形成。
DFT计算显示一个大约两倍精力充沛的处罚还原消除氟基板与未被取代的同行相比,可能占他们的不良反应。原因尚不清楚,团队选了NAr型机制激进addition-dissociation途径,尽管这似乎是一个有争议的问题因为他们不使用机械的假设合理优化的反应。
优化反应条件是相当典型的photoredox反应,有些令人惊讶的是这些基质的有限的成功。最有趣的方面是增加了化学计量钾triflate,表明关键作用的反离子反应机理。各种溶剂和碱的反应是相当宽容的,这是非常有用的应用时更复杂的基质。
反应范围是合理和相对简单的芳基烷基酸给所有可接受的收益率,和一些伟大的反应给轻易修改N-Boc-piperidine产品氟化叔碳原子,值得进一步探索。芳基卤化物的多样性是重要的,通常包括敏感功能和N-containing杂环化合物。工作轮与几个例子很好地衬托出来了如何用于访问这个反应药物类化合物。
然而,整个出版给我留下一种不完整的感觉。而反应可以提供简单的访问一些伟大的分子,作者错过机会展示其效用。修改的产品多样化的功能的例子是一个简单的和有价值的。方面的论文也削弱它的价值——比如报告孤立收益率0.1更易反应优化的规模和范围,并且没有明显的试图理解的反应;虽然机械学习是有趣的,因为作者国家新的反应所得通过机制可能不同的讨论,研究报道的反应似乎更合适。
引用
1 H王等,ACS Catal。,2020,10,4451 (DOI:10.1021 / acscatal.0c00789)






还没有评论