



美国食品及药物管理局顾问投票反对批准生原体的aducanumab不完整的数据的基础上分析
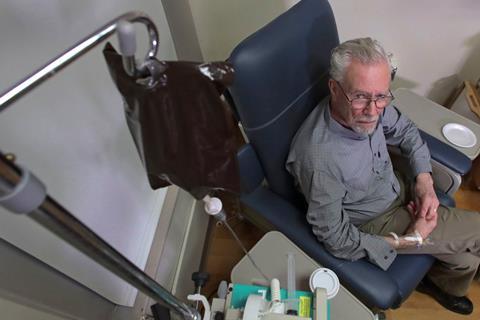
生原体周围实验性阿尔茨海默氏症的有效性的证据抗体,aducanumab,严厉批评了一个独立的专家组建议美国食品和药物管理局(FDA)。该公司已提交监管部门的批准后的治疗争议的有关临床试验停止建议可能在高剂量有效地减缓认知能力下降。
FDA内部审查出现同情生原体的临床结果的解释,但认为统计证据没有说服力。顾问小组也对分析,绝大多数投票反对推荐aducanumab批准。FDA并不一定会按照专家组的建议,并使其最终决定药物2021年3月。
有一个强有力的脱节的两个方面FDA审查,”说保罗爱森南加州大学的神经学家,我们。积极的临床评估,消极的统计评估。我还没见过。”
生原体停止招募新病人两个三期试验(称为出现和参与)2019年,临时徒劳分析后认为没有显著的临床益处的迹象。后续分析,更多的患者已经在试验的数据,建议一亚组病人在出现试验中,接受最高剂量更长时间,减少临床痴呆的分数。
生原体的申请批准是基于这一亚组分析,并提供参数为什么参与试验的效果不是很明显。FDA的评论家形容这种分析非常有说服力。相比之下,FDA生物统计学家的报告指出的全部数据似乎并不支持高剂量的效果。他继续说:“只有一个实证研究最多和第二项研究与实证研究直接冲突。顾问小组的决策支持与FDA强烈的生物统计学家,数据作为一个整体不足以支持批准该药物。
FDA的手拍了拍了这些过于协作小组成员,参与公司
阿尔茨海默氏症研究辛西娅Lemere波士顿布莱根妇女医院,我们小组的关注感到沮丧的主要目标,而“有临床效益[出现试验]全面认知和功能在每个域”。
她说,该委员会拒绝接受子群分析涉及那些收到aducanumab的高剂量。面板的看起来心烦意乱,他们被告知忽视消极[参与]试验,”当被问及评估[出现],Lemere说。大多数小组成员似乎基地试验他们的回答。”
再度重新分析临床实验数据的统计危害的方式没有最初设计到试验都记录在案。任何决定基于这样的分析应该特别仔细审查。FDA的手拍了拍了这些小组成员的协作和过于忙于公司的Lemere说。
有一件事我们可以肯定的是,这不是一个药物具有重大影响的临床疾病的进展
Lemere一直从事医学教育材料对阿尔茨海默病生原体,并在阿尔茨海默氏症协会的医学和科学咨询小组。该协会提交一份声明面板支持aducanumab批准,突出“激烈的需要提供救助和支持的数以百万计的美国人每天影响阿尔茨海默氏症的沉重现实。“Lemere预测,FDA将可能要求额外的3期研究,这可能需要4到5年。”是罕见的FDA会完全反对面板,”她说,尽管他们可以选择有条件批准,要求生原体监测患者和产生更多的数据以供稍后查看。
约翰·哈迪说,英国伦敦大学学院的许多同胞阿尔茨海默氏症的科学家他是联系难以决定是否批准为aducanumab与数据是合理的。他同情FDA的困境。尽管如此,这样的事后分析“即使可信,是危险的,”他补充道。
有一点我们可以肯定,哈代说的是,这不是一个药物具有重大影响的临床疾病的进展。“任何效应确实有可能会相对较小。尽管如此,没有其他有效治疗阿尔茨海默氏症,FDA批准aducanumab将面临压力。哈迪指出药物他克林(Cognex)阿尔茨海默病药物的一个例子,在压力下被批准的基础上可怜的审判数据和2013年撤销。
爱森说,临时分析做了一个巨大的伤害到每一个人。我们建立我们的试验给我们答复的最后审判;然后我们试图做出决定在中间,”他说。这种徒劳的分析的目的是为公司省钱和减少风险的志愿者如果试验显然是无望,但通过定义缺乏统计力量来判断小影响。我们应该更加谨慎的解释(徒劳分析),因为他们是基于不完整的数据集,”爱森说。








还没有评论