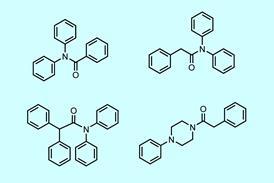


寡核苷酸药物在患者家属的压力和监管机构“以患者为中心的药物开发”的冲突中上市
经过激烈的斗争,美国食品和药物管理局(FDA)批准了首个治疗罕见且毁灭性疾病杜氏肌营养不良症(DMD)的药物。接下来是“加速审批”罗伯特·卡利夫美国剑桥制药公司Sarepta Therapeutics对Exondys 51 (eteplirsen)的科学争议进行了裁决。

Sarepta的临时首席执行官艾德·凯(Ed Kaye)宣布“患有杜氏病的男孩们将迎来伟大的一天”。然而,在此之前,FDA的一个审查小组得出结论,没有足够的证据批准Exondys 51。在什么FDA公布的文件Say也可能是第一次,简妮特FDA药物评估中心主任,驳回了审查小组的意见。
埃利斯·昂格尔,FDA药物评估办公室主任,负责审查,因此启动了内部争议程序,卡利夫支持伍德科克。安格曾警告说,如果FDA批准了Exondys 51,它将不得不批准许多其他“对绝望患者群体无效的治疗”。“这对社会和证据医学领域的损害将是巨大的,”他补充说。新闻官员桑迪·沃尔什解释说,批准是因为“FDA在评估严重和危及生命的疾病的治疗方法时,在适当的时候采取了灵活性”。
以患者为中心的药物开发是倾听患者的观点;这不是把药物批准建立在没有数据证实的轶事上
DMD几乎都发生在男性身上(大约3600分之一),它带来绝望,因为它的肌肉衰弱导致患者不幸地在年轻时死亡,通常是20多岁或30多岁。基因突变会阻止患者产生关键的肌肉蛋白营养不良蛋白,从儿童早期开始,他们逐渐失去行走和进食等能力。最终他们的心脏和呼吸衰竭。
Exondys 51是类似dna的寡核苷酸-这是第三种获得FDA批准的此类药物,针对蛋白质生产过程,这应该能使DMD患者产生缩短的肌萎缩蛋白。今年1月,FDA否决了两种作用模式相似的药物:Biomarin公司的Kyndrisa (drisapersen)——另一种寡核苷酸——和PTC Therapeutics公司的小分子药物Translarna (ataluren)。尽管Translarna已在欧洲获得有条件的批准,并在一些国家上市,但Biomarin已停止其DMD工作。
今年4月,由DMD患者家属组成的FDA情感咨询小组最初也投票反对Exondys 51。尽管患者证实服用Exondys 51后他们的健康状况有所改善,但专家组关注的是抗肌萎缩蛋白的产生。它的结论是,抗肌萎缩蛋白并没有显著增加,FDA科学家强烈批评Sarepta及其合作者进行了拙劣的实验和试验。
成本的生活
今年6月,FDA致函Sarepta,要求其提供数据,证明Exondys 51能产生具有临床意义的抗肌萎缩蛋白。当数据到达时,它确实显示了一个统计上显著的肌萎缩蛋白增加,但绝对增加非常小。在12名试验患者中,只有两名患者达到了非DMD患者1%以上的水平。昂格尔和他的同事们得出结论,这种方法不太可能带来临床益处。然而伍德科克批准了“加速审批”。这使得Sarepta能够在市场上推广这种药物,同时要求进行更多的研究来证实它的有效性。伍德科克的理由包括担心公司可能会耗尽资金。
在对伍德科克的决定提出上诉时,安格警告说,患者家属对FDA的“政治压力甚至恐吓”。他引用了数千封电子邮件,其中一些充满激情,甚至到了辱骂的地步。他还强调了FDA是如何认真考虑患者的观点的。“但以患者为中心的药物开发是要倾听患者对什么对他们重要的看法;他补充说,这不是基于没有数据证实的轶事性证词来批准药物。
然而,家庭的困境确实影响了伍德考克的思想。FDA文件援引她的说法称,如果eteplirsen等病例没有获得批准,患者就会放弃对这类产品获得批准的所有希望,从而陷入“自我治疗”的境地。
Hafedh哈达德咨询和合同研究公司B2G Life Sciences的首席执行长说,药厂对罕见“孤儿病”的批准是一个“显著的里程碑”,否则这些病是制药公司不会采用的。他说,FDA的行动清楚地证实了其“进一步加快孤儿药上市审批”的意图。



暂无评论