
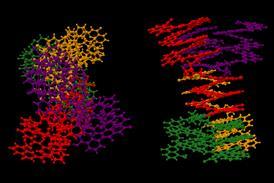


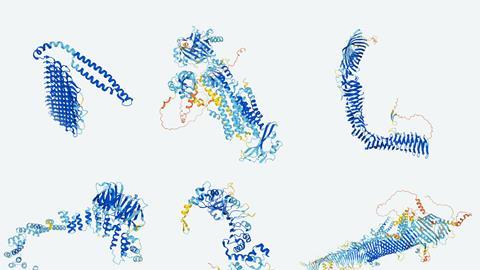

科学领域从药物发现到塑料回收后已经改变了人工智能(AI)的蛋白质结构预测研究人员共享一个巨大的数据集和开放他们的方法。7月22日,伦敦,英国DeepMind谷歌分支和欧洲分子生物学实验室(EMBL)公布的350000蛋白质结构计算其AlphaFold系统。这些包括所有20000个蛋白质从人类蛋白质组,其次是1亿多人。
AlphaFold团队强调,这是“几乎所有已知的蛋白质测序科学”。目前的估计表明,2亿或更多的蛋白质结构存在于自然。他们分子机器是单长链的氨基酸构建块折叠成独特的三维形状。知道这些形状是至关重要的对于理解蛋白质是如何工作的。
黛米斯,DeepMind的创始人兼首席执行官,描述这是人工智能”最重要的贡献了推进科学知识更新的。哈萨比斯说,它将应用程序从疾病的药物发现,蛋白质设计,理解和酶的设计”。AlphaFold已经帮助工程师快速酶回收一次性塑料,他补充说。
“对我来说,这个数据集很像人类基因组,”评论伊万伯尼EMBL的副总经理,指出资源将使以前不可能的科学。我非常激动的开始走这条路。”
DeepMind宣布之后AlphaFold团队1和大卫•贝克的RoseTTAFold集团2美国西雅图华盛顿大学的描述他们的系统和他们的代码公开可用的7月15日。2020年12月,AlphaFold赢得了蛋白质结构预测的竞争被称为结构预测的关键评估(Casp14)的挑战。然而,DeepMind没有透露它的系统是如何工作的。
细节了
RoseTTAFold团队试图发展自己的蛋白质预测工具,描述他们如何建立在五提示从DeepMind在他们的研究。”就像你听说过一个伟大的绘画和试图复制它没有任何细节,喜欢的颜色或者他们用什么方法,”华盛顿大学的计算化学家说Minkyung门敏领导RoseTTAFold项目。
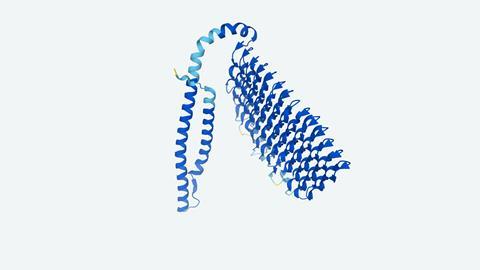
两个团队的架构都首先收集蛋白质序列数据从遗传信息通过多序列比对(MSA),这是一个过程,收集所有已知序列的进化相关感兴趣的蛋白质。
DeepMind AI提要MSA和表的配对信息到一个神经网络被称为Evoformer,组织数据和分组相似的蛋白质。它也投入学习相关的蛋白质结构称为模板。然后系统通过Evoformer输出到另一个神经网络的结构模块,来预测目标蛋白质的三维形状。同时,它估计比真正的蛋白质结构预测结构的准确性。AlphaFold不断提要Evoformer和结构模型的输出回开始的过程,直到这些估计不再提高。
相比之下,RoseTTAFold依赖三个神经网络,组织成独立的痕迹。第一个MSA不断改进。第二个做同样的对交互信息来源于MSA和已知蛋白质结构的模板。这些并行运行,每个反馈改善预测。最初以这种方式优化预测,RoseTTAFold说第三个structure-generation轨道,也反馈前两个的踪迹。让整个网络集体思考序列之间的关系,两两距离和3 d形状。
AlphaFold和RoseTTAFold可以预测蛋白质的结构与大约400个氨基酸在几分钟内。
这些工具有多好?
西蒙Erlendsson生物化学家在英国剑桥大学医学研究委员会分子生物学实验室,说研究世界是“沸腾”的新工具和他们的预测。AlphaFold和RoseTTAFold代表巨大的突破,无疑将指导结构生物学和复杂的蛋白质设计多年来,”他说。
Erlendsson研究大脑蛋白质的结构研究人员还没有完全弄明白,像弧线,帮助调节记忆。他指出,AlphaFold的预测表明,弧可能会与自己没有见过的。我真的有点不好意思,我没看到这首先,“Erlendsson说。但也许不是,对吧?”
他提出了一个关键的问题是如何好这些蛋白质的预测。在Casp14 AlphaFold中等精度的蛋白骨干在一个碳原子的宽度。然而,约翰跳投,DeepMind AlphaFold领导,指出,该系统满足三个条件时效果最好。必须有一个蛋白质的序列信息,至少30相关序列的结构是未知的,和一些相关的序列与已知的结构。精度“大幅下降”这些输入较弱时,他说。
另一个潜在问题是,蛋白质可以采用不同的形式,而且这些预测模型可以提供更有趣的活动结构。例如,g蛋白耦合的受体——“锁”在我们的细胞,引发化学反应键的变化——活跃的和不活跃的形式。确保RoseTTAFold输出活动形式在这种情况下,门敏解释说,她只是让它学习活动形式的已知蛋白质结构的模板。
尽管有这样的潜在问题,凯瑟琳Tunyasuvunakool资深研究科学家DeepMind说,AlphaFold已经可以帮助物理结构的研究。“有时当有人获得一个x射线晶体学数据集他们不能立即确定蛋白质的原子结构,”她解释说。“它有助于有一个很好的初始估计的结构。
尽管这些新技术蕴含着巨大的希望,贝克认为它们没有完全蛋白质折叠问题回答。蛋白质折叠问题的如果你的意思是预测蛋白质的结构从它的主要序列,那么这些方法接近一个解决方案,”他说。然而,从本质上说,他们是做模式识别。他们不描述蛋白质的过程从一个扩展链折叠结构。
引用
1 J跳投等,自然,2021,DOI:10.1038 / s41586 - 021 - 03819 - 2
2米门敏等,科学,2021,DOI:10.1126 / science.abj8754





还没有评论