
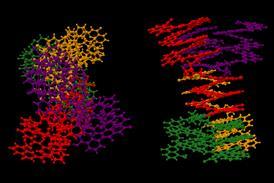


预测蛋白质结构并不一定表示函数

不久以前,一个列表的“化学的圣杯”最近编制的必威体育 红利账户可能很有可能包括“解决蛋白质折叠问题”。人们普遍相信的能力来预测蛋白质的结构从其氨基酸序列的生命科学的巨大价值。
在12月,许多媒体头条宣布了这一目标的实现。人工智能公司DeepMind表明他们AlphaFold深度学习算法可以预测蛋白质结构从他们的序列与一个原子级精度往往与获得最好的晶体分析。它被誉为一个重大突破。“它将改变一切”,进化生物学家安德烈领袖告诉自然,而结构生物学家珍妮特·桑顿说,提前将真正帮助我们理解人类如何运作和功能”。一些报道让我们相信治疗疾病,如阿尔茨海默氏症(源于蛋白质错误折叠)现在指日可待。
但这样的断言一直有争议。一些生物化学家指出,预测的准确性并不总是那么令人印象深刻,一般不太可能被接受没有实验证实,晶体学、核磁共振研究或低温电子显微镜。虽然大多数的预测结构在实验决议,不能告诉一个先天的哪些是,哪些不是,所以你需要实验来检查。也还不清楚,精度满足所需,说,发现候选药物,可以绑定到蛋白质的活性部位阻止它的功能。
解体的主要问题
别人带着问题的方法解决了蛋白质折叠问题的。基督教Anfinsen开创性工作以来,在1950年代,它已经知道解开(变性)蛋白质分子可能恢复其构象自发“土著”,这意味着仅肽序列编码的规则正确折叠。面临的挑战是找到这些规则和预测折叠路径。
AlphaFold并没有这样做。它说没有折叠的机制,只是预测结构使用标准的机器学习。它发现序列之间的相关性和结构被训练在170000左右的已知结构的蛋白质数据基础:算法不解决蛋白质折叠问题逃避它。如何“原因”从序列结构仍然是一个黑盒。
如果认为这是作弊,这并不多的实用目的。它肯定是有价值的演绎甚至是一个很好的猜测从序列结构。从我们经常可以推断蛋白质的功能和作用方式的化学机理。和“足够好”的预测是一个有用的起点与晶体数据细化。
但蛋白质折叠问题的理解的关键基因序列决定细胞功能看起来就不那么引人注目的比几十年前。我们知道实际情况要复杂得多,原因有很多。
纠缠
有更多的比正确的折叠酶作用。许多蛋白质化学修饰后在核糖体翻译:部分肽链交联,和non-amino-acid团体如卟啉或金属离子结合起来。此外,了解结构本身不会告诉你的函数。有时候这可以通过类比推导,或者说,同源性:相似的蛋白质折叠可能有类似的功能。但这并不是总是如此:以非常类似的蛋白质结构可以以化学方式截然不同,而不同的折叠可以实现类似的转换。没有独特的结构关系。
更重要的是,设计一个蛋白质配体可以挑战即使你知道它的结构非常准确,部分原因是因为我们不知道所有的规则识别——一些依赖,例如,在细节的溶剂化作用的活性部位。上游的最大障碍和药物发现通常是一个潜在的分子识别的目标——不仅因为它往往被证明是错误的目标。
在任何情况下,蛋白质功能的图片是由一个独特的和静态的晶体结构是已知的现在是过于简单化了。动力学可能是至关重要的。配体结合通常涉及一些灵活性和适应的活性部位,但更普遍的是,新兴的蛋白质功能调用系综的构象可访问:统计人口和占用时间的不同动态状态可以达到。更重要的是,许多蛋白质没有明确的折叠构象,但包含内在无序,软盘的部分肽链。这不是自然马虎:似乎功能造成障碍和灵活性。人工智能方法很可能识别哪些序列可能是无序的,但这本身不能帮助理解他们的行为。
最后,任何深度学习系统只有主管训练集的范围内。我们不知道人类蛋白质组的大小,但一些估计,只有大约5%的人类蛋白质结晶和结构决定的。因此,训练数据可能会偏向于结构相对容易解决。一些研究者认为可能会有一个系统的蛋白质结构,我们只是不知道。
这并不是贬低AlphaFold的成就——事实上我们可以预料到人工智能的方法可以帮助解决一些这些警告。真正的问题是我们很久以前就不得不放弃了这个简单的概念,任何分子的细胞的秘密是数字编码序列。
不久以前,一个列表的“化学的圣杯”最近编制的必威体育 红利账户可能包括“解决蛋白质折叠问题”。人们普遍相信的能力来预测蛋白质的结构从其氨基酸序列的生命科学的巨大价值。
在12月,许多媒体头条宣布了这一目标的实现。人工智能公司DeepMind表明他们AlphaFold深度学习算法可以预测蛋白质结构与序列通常具有原子级精度与获得最好的晶体进行了分析。“它将改变一切”,进化生物学家安德烈领袖告诉自然,而结构生物学家珍妮特·桑顿说,提前将真正帮助我们理解人类如何运作和功能”。一些报道让我们相信治疗疾病,如阿尔茨海默氏症(源于蛋白质错误折叠)现在指日可待。
但这样的断言一直有争议。一些生物化学家指出,预测的准确性并不总是那么令人印象深刻,一般不太可能被接受没有实验证实,晶体学、核磁共振研究或低温电子显微镜。也还不清楚,精度满足所需,说,发现候选药物,可以绑定到蛋白质的活性部位阻止它的功能。
别人带着问题的方法解决了蛋白质折叠问题的。基督教Anfinsen开创性工作以来,在1950年代,它已经知道解开(变性)蛋白质分子可能恢复其构象自发“土著”,这意味着仅肽序列编码的规则正确折叠。面临的挑战是找到这些规则和预测折叠路径。
AlphaFold并没有这样做。它说没有折叠的机制;如何“原因”从序列结构仍然是一个黑盒。
如果认为这是作弊,这并不多的实用目的。它肯定是有价值的演绎甚至是一个很好的猜测从序列结构。从我们经常可以推断蛋白质的功能和作用方式的化学机理。和“足够好”的预测是一个有用的起点与晶体数据细化。
但是有更多的比正确的折叠酶作用。许多蛋白质化学修饰后在核糖体翻译:部分肽链交联,和non-amino-acid团体如卟啉或金属离子结合起来。此外,知道结构本身不会告诉你功能:以非常类似的蛋白质结构可以以化学方式截然不同,而不同的折叠可以实现类似的转换。没有独特的结构关系。
更重要的是,设计一个蛋白质配体可以挑战即使你知道它的结构非常准确,部分原因是因为我们不知道所有的规则识别——一些依赖,例如,在细节的溶剂化作用的活性部位。上游的最大障碍和药物发现通常是一个潜在的分子识别的目标——不仅因为它往往被证明是错误的目标。
在任何情况下,蛋白质功能的图片是由一个独特的和静态的晶体结构太简单。动力学可能是至关重要的。配体结合通常涉及一些灵活性和适应的活性部位,但更普遍的是,新兴的蛋白质功能调用系综的构象可访问:统计人口和占用时间的不同动态状态可以达到。更重要的是,许多蛋白质没有明确的折叠构象,但包含内在无序,软盘的部分肽链。这不是自然马虎:似乎功能造成障碍和灵活性。人工智能方法很可能识别哪些序列可能是无序的,但这本身不能帮助理解他们的行为。
最后,任何深度学习系统只有主管训练集的范围内。一些人估计说,只有大约5%的人类蛋白质结晶和结构决定的。因此,训练数据可能会偏向于结构相对容易解决。一些研究者认为可能会有一个系统的蛋白质结构,我们只是不知道。
这并不是贬低AlphaFold的成就——事实上我们可以预料到人工智能的方法可以帮助解决一些这些警告。真正的问题是我们很久以前就不得不放弃了这个简单的概念,任何分子的细胞的秘密是数字编码序列。





还没有评论