



算法可能会学到关于能源景观,但它需要一点帮助找到全球最低
的成功AlphaFold超过2亿种蛋白质的结构预测,宣布去年8月,导致兴奋的说算法将彻底改变生物学、药物发现和分子医学。这还有待观察,但一些人渴望脾气的炒作指出AlphaFold没有事实上解决了蛋白质折叠的问题。相反,它回避了这个问题通过使用机器学习来找到序列和已知结构之间的关联,然后全面未知结构。
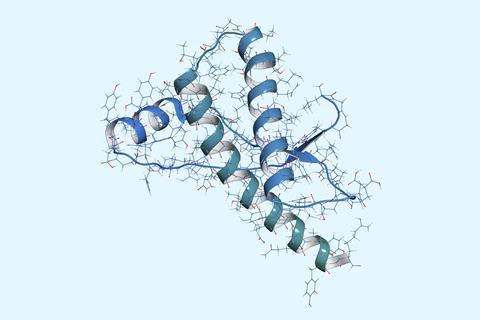
不同,分子动力学模拟中,在DeepMind AlphaFold——由一个团队,谷歌的一个分支,并不试图概括的分子途径导致折叠结构。它只是使用序列之间的相关性,获悉和链的形状。识别这些对于一个任意的主要氨基酸序列,然而,该算法需要编译的集合序列密切相关的目标序列,称为多重序列比对(MSAs)。这些给算法的氨基酸替换的后果是这部分的配置空间。这要求MSAs是一个问题如果有几个已知的蛋白质同系物接近目标。
这些预测有多好仍然有争议。一个新的预印本报告说,即使对于结构AlphaFold预测高信心,可以有重大差异的细节与实验数据。1预测,研究人员说,应该不是视为替代实验结构的决心,但相反,假设实验可以测试。
不过,预测往往是令人印象深刻的准确和肯定有用的探索蛋白质的结构和功能。不过,他们告诉我们任何关于蛋白质折叠本身?
地面控制
认为蛋白质的最低折叠态独特编码在其主要氨基酸序列在1973年提出的基督教Anfinsen。2蛋白质还能如何正确、可靠地折叠核糖体一旦被翻译,或变性后再折起?今天的标准图片,折叠发生在势能与烟囱似的地形景观,确保过程是引导到基态或多或少无论初始配置是什么,不困在次优亚稳构象。
AlphaFold告诉我们任何关于这个能源格局吗?并不明显,但哈佛大学的詹姆斯·罗尼和Sergey Ovchinnikov现在认为它确实是这样。3他们建议的算法具有隐式学习能源格局的样子,但是,因为它是如此巨大,它不能导航的全球能量最低最稳定的褶皱——从任意起点。这就是为什么需要MSAs:启动优化过程正确的附近。(“能源”在这里并不一定对应热力学自由能,而是一些函数AlphaFold优化)。
支持这张照片,罗尼和Ovchinnikov表明AlphaFold可以推断序列符合候选人如何结构提供了“手工”。蛋白质结构的研究人员使用一个数据库开发使用古典结构预测软件包罗塞塔。4罗塞塔有一个显式的能量函数,基于分子间作用力,溶剂化作用,氢键等等,对小型蛋白质并执行得很好。通过喂养AlphaFold结构优化罗塞塔连同许多“诱饵”结构涵盖了广泛的结构空间,研究人员发现,信心算法分配给每一个结构与他们真正的质量而言,如何最小化能量函数。
他们使用一组相似的目标和诱饵的发达14日结构预测的关键评估挑战2020年,工作两年一次的“比赛”中,彼此对抗的预测方法。这里太AlphaFold需要诱饵——实际上,充当MSAs——得到的轴承产生好的CASP14的预测结构的目标。
现在看起来更少的限制,。鉴于AlphaFold似乎得到表面局部能量的感觉,它可能不是必要从真正的蛋白质来帮助它收集MSAs导航到全球最低。方法像罗塞塔可以从头生成足够合理的结构对于一个给定的序列可能足够AlphaFold其中最好的识别和改善。这是特别好的消息使用算法来设计全新的蛋白质。这个人工智能,甚至比看起来更聪明:编码在其信心估计是一种直觉的物理学。
引用
1 T C Terwilliger等,bioRxiv,2022,DOI:10.1101 / 2022.11.21.517405
2 C B Anfinsen,科学,1973,181年,223 (DOI:10.1126 / science.181.4096.22)
3 J P罗尼和S Ovchinnikov,理论物理。启。,2022,129年,238101 (DOI:10.1103 / PhysRevLett.129.238101)
4 R F阿尔弗德等,j .化学理论第一版。,2017,13,3031 (DOI:10.1021 / acs.jctc.7b00125)











还没有评论